





La pandemia e il ruolo chiave del sequenziamento genomico
A metà gennaio del 2020 un gruppo di ricercatori cinesi e australiani pubblicava per la prima volta l’intera sequenza di quel virus, allora sconosciuto, che stava provocando un esteso focolaio di casi di polmonite ad eziologia non nota nella città di Wuhan. Il patogeno, che sarebbe poi stano denominato SARS-CoV-2, cominciava ad assumere un’identità molto più precisa: il suo lungo genoma, composto da quasi 30 mila basi, era stato decifrato e si iniziava a conoscere meglio anche la sua formidabile chiave di accesso alle cellule umane, quella proteina spike che è in grado di legarsi in modo estremamente efficace al recettore Ace2, presente in modo diffuso nell’intero organismo umano e soprattutto nelle vie respiratorie.
Come tutti i virus a RNA anche SARS-CoV-2 è soggetto a cambiamenti. Riproducendosi copia il suo materiale genetico e in questo processo è facile che si manifestino delle alterazioni. Pur essendo molto più stabile rispetto ad altri virus, grazie a un enzima che funge da riparatore degli errori, SARS-CoV-2 è dunque in continua evoluzione e sebbene la maggioranza delle mutazioni non implichi conseguenze biologiche rilevanti è sempre possibile che il patogeno riesca a conservare quelle alterazioni che vanno a suo vantaggio.
Lo stiamo vedendo proprio in questi giorni: le varianti inglese, sudafricana e brasiliana sembrano più facilmente trasmissibili, come anche il ceppo D614G, protagonista della prima ondata pandemica che ha investito l’Italia e l’Europa in primavera. Un contagio che galoppa veloce è indubbiamente un punto di forza per il virus perché lo porta a trovare con più facilità nuovi ospiti da infettare. Una mutazione che si traducesse in una malattia più grave non sarebbe invece un vantaggio. Indubbiamente non lo sarebbe per noi, ma nemmeno per il virus perché una delle sue “strategie” più brillanti è quella di riuscire a propagarsi efficacemente attraverso le persone asintomatiche o paucisintomatiche, cosa che la SARS, caratterizzata da un decorso clinico molto più grave, non aveva potuto fare.
Allo stesso modo acquisire la capacità di nascondersi dal sistema immunitario, mettendo così a rischio l’efficacia dei vaccini e degli anticorpi sviluppati dalle persone guarite dall’infezione, migliorerebbe le possibilità del virus di restare tra noi più a lungo.
Dall’inizio della pandemia, a dicembre erano già oltre 4000 le sole mutazioni che interessano la proteina spike del virus e a livello mondiale il principale sforzo nel lavoro di sequenziamento genomico è quello effettuato dal Consorzio britannico COG-UK che ha contribuito per circa la metà degli oltre 400.000 genomi di SARS-CoV-2 sequenziati e archiviati su GISAID, un database online senza scopo di lucro per la condivisione di genomi virali. All’archivio contribuiscono più di 140 Paesi ma la maggior parte ha caricato solo un piccolo numero di sequenze.

Il 10 gennaio 2020 China CDC condivideva su GISAID il primo sequenziamento genomico di SARS-CoV-2 dando inizio al percorso che ha portato alla sviluppo dei test diagnostici e dei vaccini. Immagine da GISAID
Eppure seguire l’evoluzione del virus e capire come cambia è fondamentale e probabilmente lo sarà sempre di più anche in futuro perché la pressione dei vaccini potrebbe indurre SARS-CoV-2 ad aumentare la sua variabilità, cercando nelle mutazioni una strada per la sopravvivenza.
Il 2021, mette in guardia un articolo di Nature, si preannuncia come l’anno della varianti. Negli ultimi due mesi sono stati rilevati più frequentemente nuovi ceppi e, dal momento che la sorveglianza del genoma virale è irregolare a livello globale, non si può escludere che altre varianti potenzialmente preoccupanti siano già in circolazione. Tra i Paesi che hanno avuto meno successo nel far funzionare l'epidemiologia genomica, approfondisce l’articolo di David Cyranosky, ci sono gli Stati Uniti che hanno condiviso su GISAID meno dello 0,3% del numero totale di infezioni da SARS-CoV-2. In termini assoluti il Regno Unito ha caricato sul database il numero maggiore di sequenze: secondo l’ultimo report del consorzio COG-UK sono oltre 200 mila. Al secondo posto si colloca la Danimarca che ha contribuito al 7% dell'archivio. Se si considera invece la percentuale di sequenze analizzate rispetto al totale dei casi di contagio confermati in un determinato paese in testa c'è l'Australia che raggiunge addirittura il 60% (su un totale molto limitato di infezioni, visto che dall'inizio della pandemia il numero di persone che hanno contratto il virus è inferiore a 30 mila).
Nei giorni scorsi sia l'Organizzazione mondiale della sanità che il Centro europeo per il controllo delle malattie hanno sottolineato la necessità di espandere il monitoraggio delle varianti emergenti di SARS-CoV-2.
Il 12 gennaio il tema è stato al centro di una riunione virtuale convocata dall'Oms a cui hanno preso parte più di 1.750 scienziati di 124 Paesi del mondo. Maria Van Kerkhove, responsabile tecnico dell'Oms per Covid-19 ha ricordato che la maggior parte delle sequenze genetiche pubblicate "proviene solo da una manciata di paesi. Migliorare la copertura geografica del sequenziamento è fondamentale affinché il mondo abbia occhi e orecchie sui cambiamenti del virus". Gli scienziati hanno sottolineato l'importanza delle piattaforme di dati nazionali per documentare dati clinici ed epidemiologici che facilitino la valutazione dei ceppi virali emergenti e c'è stato consenso sulla necessità che il monitoraggio sia accompagnato da una tempestiva condivisione di campioni virali, tramite meccanismi concordati a livello globale, in modo che la ricerca possa essere avviata prontamente ogni volta che è necessario.
L'Ecdc ha poi mandato indicazioni precise ai laboratori europei con l'obiettivo di capire quanto sia realmente diffusa la variante inglese. Il documento spiega che "è cruciale continuare con la sorveglianza, anche con una raccolta mirata di campioni, per rilevare la presenza di varianti" e, in accordo con i suggerimenti dell'Oms, si raccomanda di dare priorità nel sequenziamento anche alle persone che hanno ricevuto il vaccino anti-Covid e poi si sono contagiate, a chi lavora in strutture a rischio, dove si ha una stretta interazione con animali suscettibili al virus, o dove ci sono pazienti immunodepressi, specialmente se ricevono una terapia a base di anticorpi monoclonali. Altre situazioni che richiedono particolare sorveglianza sono il verificarsi di un cambiamento o di aumento inaspettato nella trasmissibilità o virulenza e quando si sospetta una diversa resa delle terapie usate. Oltre a questa lista, vanno anche monitorati i contagi collegati a viaggi in aree a rischio.
La preoccupazione degli esperti per la diffusione della variante inglese B.1.1.7 è condivisa e guardando ai dati che arrivano dal Regno Unito vi è ragione di crederlo: nell'ultima settimana i contagi, pur in diminuzione dopo le rigide misure restrittive introdotte dal governo, sfiorano ancora i 300 mila casi e il 20 gennaio il Paese ha fatto registrare 1820 decessi giornalieri, un numero che non era mai stato raggiunto in precedenza. La variante non è caratterizzata da una maggiore patogenicità ma essendo facilmente trasmissibile porta ad un significativo aumento dei contagi e, di conseguenza, anche dei casi con decorso clinico grave.
In questa fase, oltre a mantenere le misure di contrasto alla pandemia e a procedere con le vaccinazioni, diventa centrale avere la capacità di monitorare efficacemente la circolazione della variante inglese (e delle altre varianti sotto osservazione, come quella sudafricana e brasiliana) per isolarla in modo tempestivo ed impedirne l'ulteriore diffusione. Un'attività che però in Italia, e in larga parte degli altri Paesi europei, non viene effettuata in maniera estesa. Anche gli Stati Uniti, approfondisce Nature, hanno avuto difficoltà nel far funzionare l'epidemiologia genomica nonostante a maggio fosse stato lanciato un programma appositamente dedicato, chiamato SPHERES. Lo sforzo non si è evoluto in un sistema nazionale e la maggior parte del sequenziamento è effettuata dai singoli laboratori accademici più che dai grandi centri genomici del paese.
Secondo i dati raccolti dal Global Report Investigating Novel Coronavirus Haplotypes, basati sulle informazioni disponibili sulla piattaforma GISAID, anche in seguito alla scoperta della variante B.1.1.7 la sorveglianza molecolare è rimasto limitata, ad eccezione del Regno Unito e della Danimarca che hanno sequenziato rispettivamente 107.306 e 19.938 campioni virali. In Italia è stato analizzato il genoma virale di appena 467 persone e 82 di queste sono risultate positive alla variante inglese.
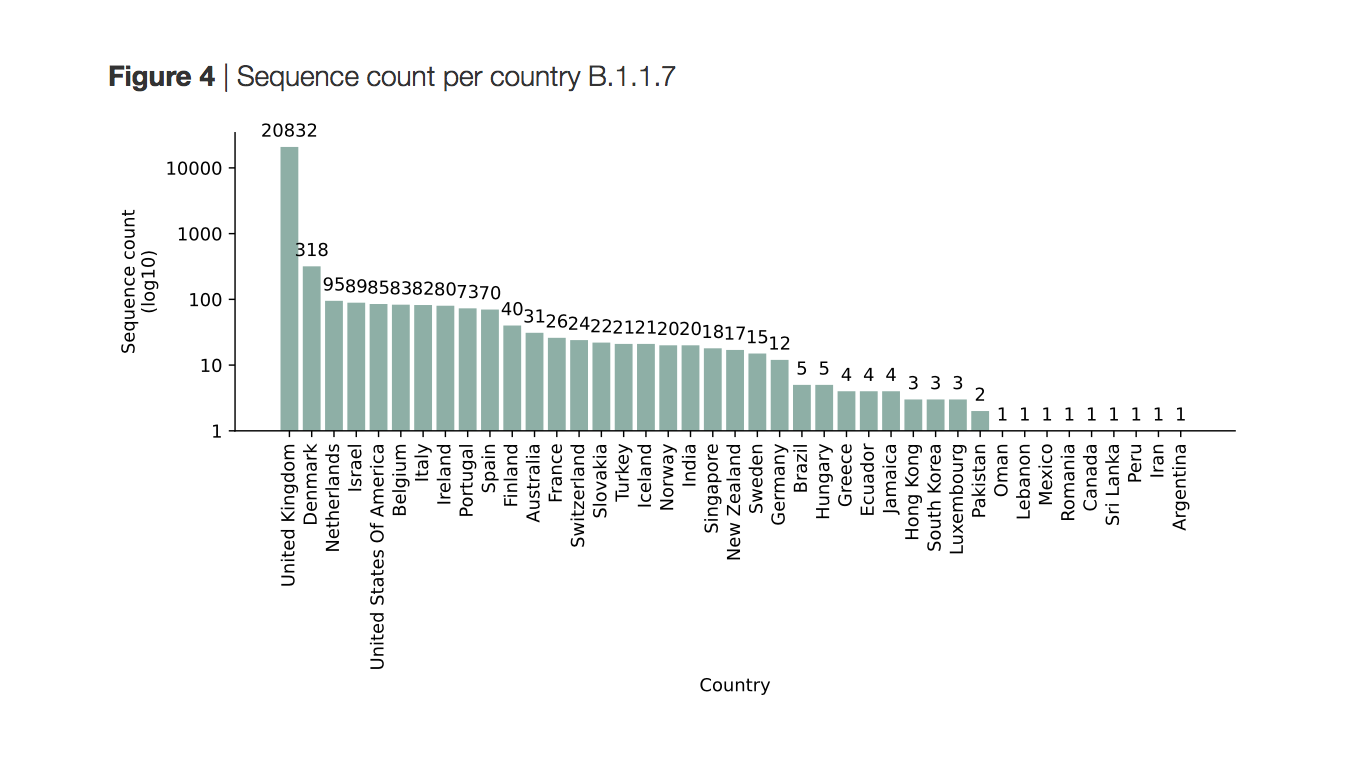
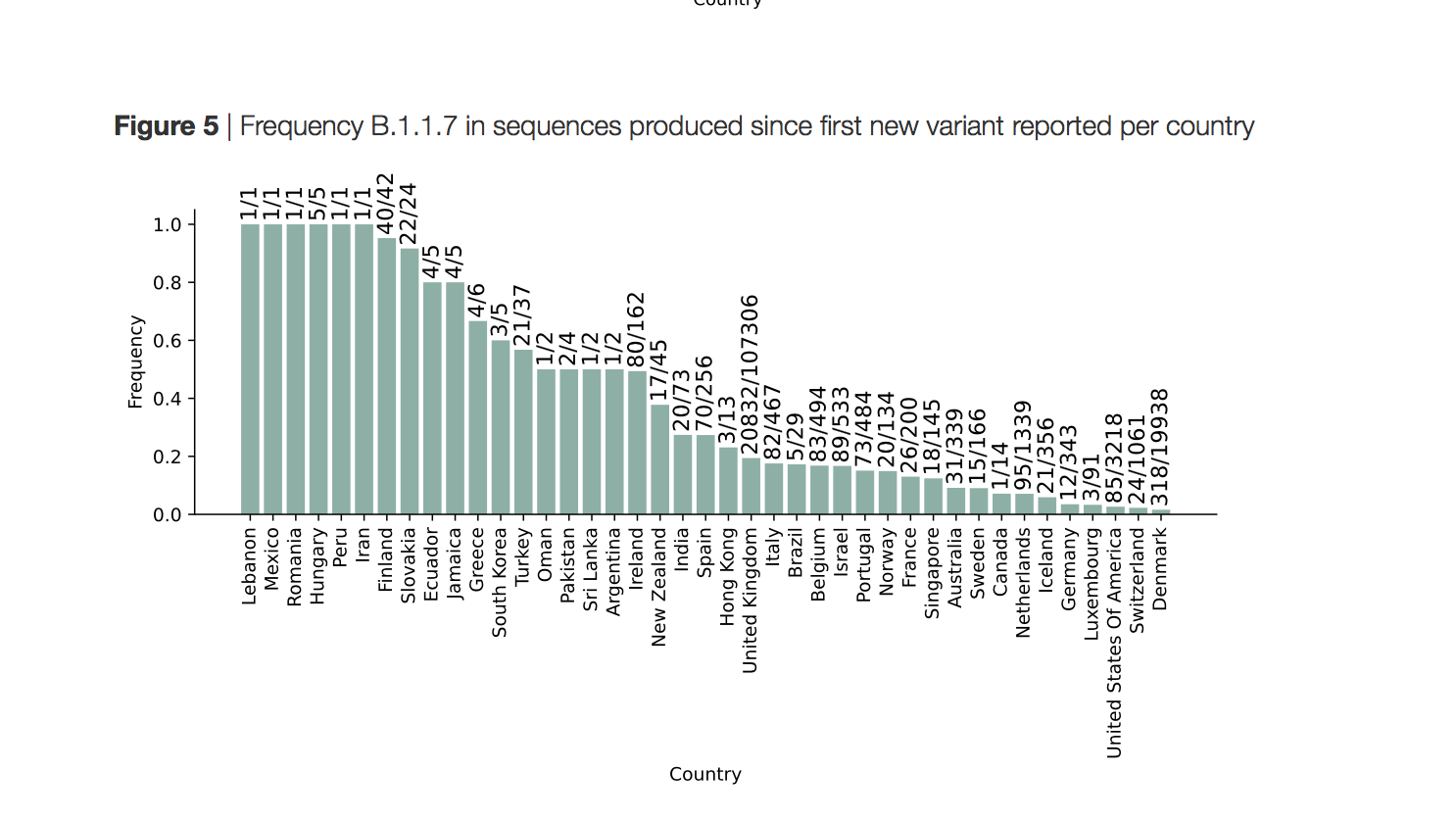
La prima figura riporta il numero totale di sequenze virali identificate come B.1.1.7 in diversi paesi. La seconda mostra la frequenza di B.1.1.7 rispetto al totale del sequenziamenti effettuati. Grafici tratti da GRINCH
Abbiamo chiesto a Marco Gerdol, ricercatore di Genomica all'università di Trieste, come andrebbe impostato un programma di sorveglianza molecolare più esteso e se, al momento attuale, è possibile sapere con sufficiente certezza quanto la variante inglese sia già diffusa in Italia.
Intervista a Marco Gerdol, ricercatore di Genomica dell'università di Trieste, sulla sorveglianza molecolare del virus SARS-CoV-2. Servizio e montaggio di Barbara Paknazar
"Non abbiamo modo di sapere con sufficiente certezza quanto sia diffusa in Italia la variante inglese perché la sorveglianza molecolare, almeno finora, è stata lasciata all’iniziativa dei singoli", spiega Marco Gerdol, ricercatore di Genomica all'università di Trieste, aggiungendo che, più in generale, "manca un programma di coordinamento nazionale finalizzato a una raccolta dei dati che sia rappresentativa delle diverse varianti in circolazione nel nostro paese. Lo stesso accade in quasi tutta Europa, ad eccezione di Regno Unito e Danimarca, perché probabilmente questa attività non è stata ritenuta strategica ai fini del contenimento della pandemia".
Gerdol specifica che a mancare non sono le strutture. "Abbiamo diverse realtà d’eccellenza in Italia che negli ultimi anni si sono dotate dei mezzi necessari e offrono tuttora, sia nel pubblico che nel privato, dei servizi di sequenziamento di vario tipo e che potrebbero essere destinati a questo genere di sorveglianza". Il problema è da individuare più nell'assenza di un coordinamento a livello centrale. "Mancando un modo di coordinare queste risorse non esiste una strategia comune e i singoli laboratori agiscono un po’ di testa propria, senza grande comunicazione tra loro e con gli organi nazionali che dovrebbero essere deputati a questo coordinamento".
L'assenza di una regia comune e standardizzata, spiega il ricercatore, ha spesso indirizzato le attività di sequenziamento verso "l'analisi dei casi ritenuti interessanti, magari per perché avevano particolari risvolti clinici: il rischio è anche quello di andare a descrivere singoli casi particolari e ritenere che siano rappresentativi di una situazione generale. Lo abbiamo visto anche durante l’estate quando si susseguivano notizie relative a presunte varianti virali meno pericolose, pur nella consapevolezza che questa evoluzione va in direzione opposta alle aspettative legate alla selezione naturale".
Ed è lo stesso meccanismo che sta caratterizzando anche queste settimane in cui il lavoro di rilevamento della variante B.1.1.7 si è intensificato. "Nell’ultimo periodo c’è stato un sampling bias nei confronti della variante inglese perché ci si è concentrati nell’investigare pazienti sintomatici che avessero una storia molto recente di viaggio nel Regno Unito. Se guardiamo alle sequenze genomiche depositate dall’Italia negli ultimi giorni si rileva un numero molto rilevante di genomi sequenziati in Abruzzo, dove è stato fatto un buon lavoro ma con un forte bias proprio verso i genotipi riconducibili alla variante inglese perché è stato individuato un focolaio piuttosto significativo in provincia di Chieti. Questo non è il modo in cui dovrebbe essere svolta una sorveglianza molecolare attendibile", approfondisce Marco Gerdol.
Pochi dubbi sul modello a cui ispirarsi. "A mio avviso è sicuramente quello inglese. Un network nazionale di molti laboratori che è stato possibile grazie alla presenza di infrastrutture già preesistenti, come il Sanger Institute, ma anche grazie ad una forte partecipazione del governo a livello economico", fa notare il ricercatore dell'università di Trieste.
In questi mesi di pandemia il Consorzio COVID-19 Genomics UK (COG-UK) è diventato il punto di riferimento a livello mondiale per il sequenziamento, rapido e su larga scala, del genoma di SARS-CoV-2. Istituito alla fine dl marzo 2020 con un finanziamento di 20 milioni di sterline da parte del Dipartimento per la salute e l’assistenza sociale del Regno Unito (Dhsc), Uk Research and Innovation (Ukri) e Wellcome Sanger Institute, COG-UK raggruppa centri di ricerca e università britanniche e svolge un lavoro prezioso che permette di identificare e comprendere i cambiamenti genetici messi in atto dal virus durante le sue continue replicazioni. E' presieduto da Sharon Peacock, microbiologa dell'Università di Cambridge, che in un'intervista a Nature ha spiegato come la sua idea di creare un consorzio dedicato a questo scopo fosse stata inizialmente respinta perché i coronavirus mutano meno dell'influenza e alcuni ricercatori ritenevano che gli sforzi di sorveglianza avrebbero portato a individuare solo mutazioni prive di significato.
"Il vero impatto della sorveglianza genomica diventerà chiaro quando le persone vedranno le prove che ha prevenuto malattie e morte", ha affermato Peacock e il Regno Unito ha poi compiuto un passo ulteriore costituendo il consorzio G2P-UK che ha il compito di lavorare al fianco di COG-UK per capire se le mutazioni che stanno emergendo abbiano un effetto sulla trasmissibilità del contagio, sulla gravità della forma di Covid-19 e sull'efficacia di vaccini e cure.
Tornando alle tre varianti emerse recentemente Marco Gerdol sottolinea che "l’analisi della diversità molecolare dei genotipi sequenziati ci dice, ad esempio, che la variante inglese è già entrata in Italia attraverso svariate dozzine di introduzioni, indipendenti l’una dall’altra".
Una considerazione che fa comprendere quanto "una misura come la chiusura dei confini abbia un senso limitato soprattutto nel momento in cui l’emergenza viene resa nota a diverse settimane di distanza dall’esplosione delle varianti stesse. La decisione dell’Italia e di molti altri Paesi europei di interrompere i voli da e verso il Regno Unito è soprattutto una misura dimostrativa, ma non ha avuto un gran effetto pratico dal momento che moltissimi casi erano già stati esportati. E probabilmente lo stesso è accaduto, anche se non ne abbiamo traccia, per la variante sudafricana che sappiamo circolare a bassi livelli da oltre un mese in altri paesi europei in cui si fa sorveglianza. Il medesimo discorso vale anche per quella brasiliana, anche perché Manaus è una metropoli".
Le tre varianti hanno un’origine evolutiva distinta ma hanno molti aspetti in comune, come ci spiega Marco Gerdol. "Tutte sono caratterizzate dalla presenza condivisa di una mutazione particolare, la famosa N501Y, una sostituzione non sinonima di una asparagina con una tirosina a carico del receptor binding domain della proteina spike che può avere delle importanti implicazioni nell’interazione con il recettore Ace2 delle nostre cellule. Questa mutazione è stata acquisita in modo indipendente nei tre lignaggi che sono caratterizzati anche da un numero abbastanza considerevole di mutazioni non sinonime, sempre riguardanti la proteina spike, che in alcuni casi sono condivise tra le varianti e in altri no.
Tra le tre varianti quella inglese è quella che porta delle alterazioni strutturali meno significative a carico della proteina spike. La variante sudafricana e quella brasiliana hanno invece in comune alcune mutazioni che riguardano la posizione 484 ed è già stato visto, da saggio in vitro, che mutazioni a carico di questo amminoacido sono legate a fenomeni di evasione immunitaria. Le due varianti inoltre condividono anche una terza mutazione in posizione 417 con il cambio di un amminoacido. L’interazione tra queste mutazioni è molto complessa e gli effetti sia dal punto di vista strutturale, che i risvolti immunologici, non sono di facile studio. E’ molto difficile prevedere in silico quali effetti possano provocare mutazioni che sono anche così distanti tra loro nella sequenza lineare della proteina, sia a livello di riconoscimento immunitario che di interazione tra spike e Ace2. In questi giorni gli studi in vitro stanno ovviamente andando avanti ma per le ultime varianti, descritte ai primi di gennaio, non abbiamo ancora a disposizione i risultati".
Come aveva osservato anche l'infettivologo Stefano Vella in una recente intervista a Il Bo Live, il fatto che la variante ritenuta più allarmante sia emersa a Manaus probabilmente non è frutto del caso. "E' atteso che fenomeni di fuga immunitaria possano avvenire in modo particolare in contesti dove il virus ha già circolato in modo molto significativo e quindi la frazione di individui suscettibili nella popolazione sia basso e si sia raggiunto un livello, almeno ipotetico, di immunità di comunità", concorda Gerdol.
E aggiunge: "ci sono infinite possibilità che emergano ulteriori varianti con le stesse caratteristiche, magari proprio in regioni del mondo dove il virus si è già ampiamente diffuso e si è arrivati vicini a una soglia di immunità di comunità. Penso ad alcune aree dell’America del sud come Perù, Bolivia e certe parti del Messico: potrebbero essere delle bombe ad orologeria dal punto di vista dell’esplosione di nuove varianti caratterizzate da fenomeni di evasione immunitaria".
"Il mio auspicio - conclude il ricercatore di Genomica dell'università di Trieste - è che in un futuro molto prossimo le strategie di sorveglianza vengano implementate e in questa direzione va anche l’appello dei Biologi per la scienza, un gruppo di giovani biologi che si battono per queste giuste cause. Spero che la loro voce, insieme a quella di colleghi che ricoprono posizioni importanti nel panorama della ricerca italiana e internazionale, possa portare ad un cambio di rotta nella strategia italiana".


