





La nostra genetica e quella del virus

Image credits: Frontline genomics, via Biospectrum
Perché le persone rispondono così diversamente al virus?
Uno degli aspetti ancora meno compresi di CoVid-19 è che la malattia ha decorsi molto diversi in persone diverse. Alcuni di quelli che contraggono il virus non sviluppano mai sintomi (asintomatici), altri ne sviluppano di molto lievi (pauci-sintomatici); tra coloro che sviluppano la sindrome respiratoria alcuni se la cavano con qualche giorno di febbre, altri necessitano il ricovero e una parte di questi addirittura la terapia intensiva. Alcuni pazienti vanno anche incontro al decesso. Alcune risposte potrebbero risiedere nella genetica.
Uno studio uscito a giugno su New England Journal of Medicine ha analizzato i genomi di più di 1500 pazienti affetti da CoVid-19 in Italia e Spagna (e più di 2000 nel gruppo di controllo) per cercare varianti genetiche che possano favorire la suscettibilità a sviluppare forme gravi della malattia. Lo studio di associazione genomica (genome wide association study) ha confermato un dato che già si conosceva e ne ha trovato uno nuovo. Si sapeva già che il virus Sars-CoV-2 fa il suo ingresso nell’organismo tramite un recettore delle mucose respiratorie, ACE2, e chi ha più recettori di questo tipo è più soggetto all’infezione. Lo studio ha individuato sul terzo cromosoma (3p21.31) uno di questi geni, SLC6A20, che contribuisce all’espressione del recettore ACE2.
Lo studio, a prima firma di David Ellinghaus, ha trovato anche un altro gene sul cromosoma 9 (9q34) associato al rischio di forme gravi di CoVid-19 ed è quello che determina il gruppo sanguigno: in particolare gli individui A positivi sembrano quelli più a rischio, mentre i portatori del gruppo 0 sembrano più protetti. Una rianalisi successiva tuttavia, compiuta dal più ampio consorzio al mondo che sta studiando la nostra genetica in rapporto al virus, ha stabilito che le evidenze per sostenere un’associazione tra geni del gruppo sanguigno e malattia non sono forti a sufficienza. Sono stati identificati invece 12 geni coinvolti nella risposta immunitaria che regolano la produzione di citochine e che potrebbero giocare un ruolo nelle espressioni più severe della malattia, quando si scatena la tempesta citochinica che aggrava le condizioni del paziente.
Svante Pääbo, direttore del Max Planck Institute di antropologia evoluzionistica di Lipsia, che nel 2010 ha sequenziato l’intero genoma di Neanderthal e successivamente dell’uomo di Denisova, si è accorto che alcune delle varianti geniche pubblicate da Ellinghaus e colleghi sul NEJM, in particolare quelle presenti sul terzo cromosoma, sono presenti anche nei Neanderthal vissuti 60.000 anni fa in Croazia. Questo pacchetto genetico è arrivato a noi sapiens dai Neanedrthal, con i quali ci siamo più volte accoppiati in Eurasia, una volta usciti dall’Africa. Di conseguenza questo gruppo di varianti geniche di origine neanderthaliana è particolarmente diffuso in Asia (nel 30% degli abitanti), in Europa (nell’8% degli europei), ma non in Africa. Il che, se confermato (lo studio è disponibile in preprint su bioRxiv), potrebbe essere anche parte della spiegazione del perché in Africa, finora, il virus sembra aver attecchito meno che in altri Paesi.
The major genetic risk factor for severe COVID-19 is inherited from Neandertals https://t.co/vm3SKyRq6J #biorxiv_genomic
— bioRxiv Genomics (@biorxiv_genomic) July 3, 2020
Un altro lavoro che promette di restituirci informazioni importanti su quali caratteristiche genetiche umane favoriscano la suscettibilità al virus e quali invece inducono una maggiore protezione è lo studio genetico in corso a Vo’ Euganeo. Dopo aver effettuato l’analisi epidemiologica tra fine febbraio e primi di marzo e aver pubblicato i risultati su Nature, il team diretto da Andrea Crisanti è passato alla raccolta dei dati sierologici, per comprendere la risposta immunitaria degli abitanti, e dei dati genetici. Le analisi genetiche richiederanno diversi mesi e in autunno forse ne sapremo qualcosa di più.
Il virus ha sviluppato mutazioni?
Tutti i virus tendono a mutare mano a mano che infettano l’ospite. La maggior parte delle mutazioni non modificano le proprietà del virus, molte sono deleterie e dunque non vengono conservate nel tempo, mentre alcune possono risultare vantaggiose e verranno mantenute se aumenteranno il successo riproduttivo del virus. È l’evoluzione, bellezza. Tenendo traccia delle mutazioni del virus e dei suoi spostamenti in diverse aree geografiche della Terra è possibile ricostruirne la storia filogenetica.
Nell’ecosistema mediatico italiano, dopo la fine del lockdown, ha tenuto banco l’idea che il virus si fosse indebolito, dacché il numero di contagi giornaliero dai picchi di più di 6000 casi al giorno di marzo è sceso a 300 a giugno e 200 a luglio. Ad oggi non esiste in letteratura una singola evidenza che il virus, tanto meno quello italiano, abbia accumulato mutazioni che ne abbiano depotenziato l’azione infettiva. Anzi, la letteratura scientifica disponibile va semmai nella direzione opposta. Ma partiamo dall’inizio.
L’epidemia da Sars-CoV-2 parte in Cina nella provincia di Wuhan a dicembre 2019 (anche se oggi sappiamo che probabilmente il virus era presenta mesi prima, forse già ad agosto). Un gruppo di ricercatori cinesi guidato da Shi Zeng-Li a inizio febbraio pubblica su Nature la sequenza completa del virus, identico per quasi l’80% del suo genoma al suo parente del 2003, Sars-CoV, e per il 96% identico al coronavirus del pipistrello detto “a ferro di cavallo” (horseshoe bat) per la conformazione del muso. Si vede anche subito che il recettore ACE2 è la porta di ingresso del virus nell’organismo umano e che i pazienti trattati con il plasma contenente anticorpi rispondono bene alla terapia.
Da Wuhan il virus inizia a spostarsi, negli Stati adiacenti alla Cina prima e poi anche in Europa. In Germania prima (si pensa oggi) e poi in Italia, dove per la prima volta viene riconosciuto il 21 febbraio nel lodigiano in Lombardia. Il primo decesso europeo invece è un abitante di Vo’ Euganeo. Nel frattempo il virus aveva già attraversato l’oceano ed era sbarcato anche negli Stati Uniti.
Research out today in the journal Cell shows that a specific change in the SARS-CoV-2 coronavirus virus genome, previously associated with increased viral transmission and the spread of COVID-19, is more infectious in cell culture. https://t.co/rvYhWcDOgL
— Los Alamos Lab (@LosAlamosNatLab) July 3, 2020
A inizio luglio viene pubblicato uno studio sulla rivista Cell diretto da Bette Korber dei laboratori di biologia teorica e biofisica di Los Alamos. Il tasso di mutazione del virus sembra essere basso, così come sembra essere ridotta la diversità genetica di Sars-CoV-2: significa che le mutazioni accumulate sono poche e i virus in giro per il mondo si assomigliano tutti geneticamente. Tuttavia, anche questa bassa diversità secondo i ricercatori è sufficiente a permettere alla selezione naturale di avvantaggiare una variante più performante a scapito di un’altra. Quando la selezione naturale agisce lo fa favorendo le varianti di maggior successo riproduttivo. In altri termini se emerge una mutazione che rende il virus più facilmente trasmissibile, la selezione premia la sua corsa evolutiva.
Lo studio pubblicato su Cell si è concentrato sulle mutazioni della proteina Spike, quella che il virus usa come chiave da infilare nella serratura del nostro organismo (il recettore ACE2). Sul contrasto dell’azione della proteina Spike si stanno concentrando anche gli studi per lo sviluppo di un vaccino, ragione per cui è stata messa sotto i riflettori dei genetisti: una sua eccessiva propensione a mutare infatti costituirebbe un problema per la capacità di calibrare un vaccino o un farmaco efficace.
I ricercatori hanno analizzato più di 28.500 sequenze virali caricate sul database GISAID (che oggi è arrivato a contenere 63.000 sequenze genetiche del virus) e hanno trovato che la variante D614, che era la più diffusa fino a febbraio, è stata oggi quasi completamente sostituita dalla variante G614 (o D614G), comparsa in Europa e ora diffusasi in tutto il mondo, con due eccezioni: l’Islanda e la contea di Santa Clara in California.
Le prime mutazioni G614 sono state rintracciate dai ricercatori proprio in Italia intorno al 20 febbraio e poi in molti altri Paesi. In Italia, come in altri Paesi europei, l’epidemia è iniziata con entrambe le varianti (D614 e G614) ma presto, già a marzo, quella più aggressiva si è imposta sull’altra.
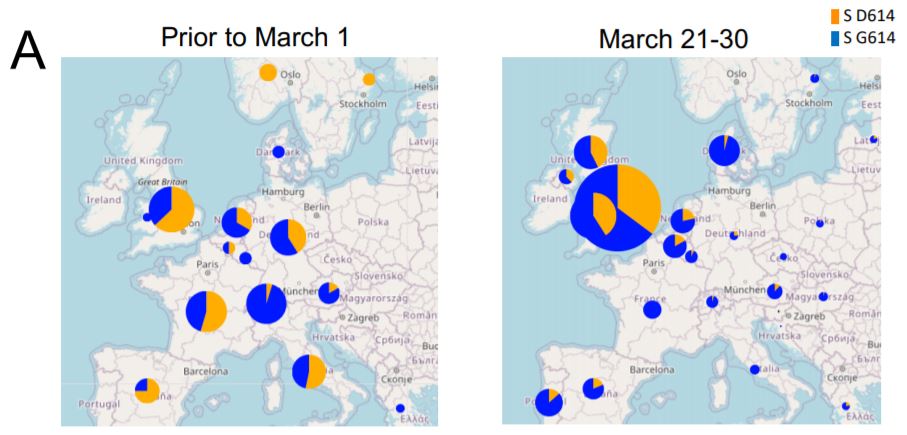
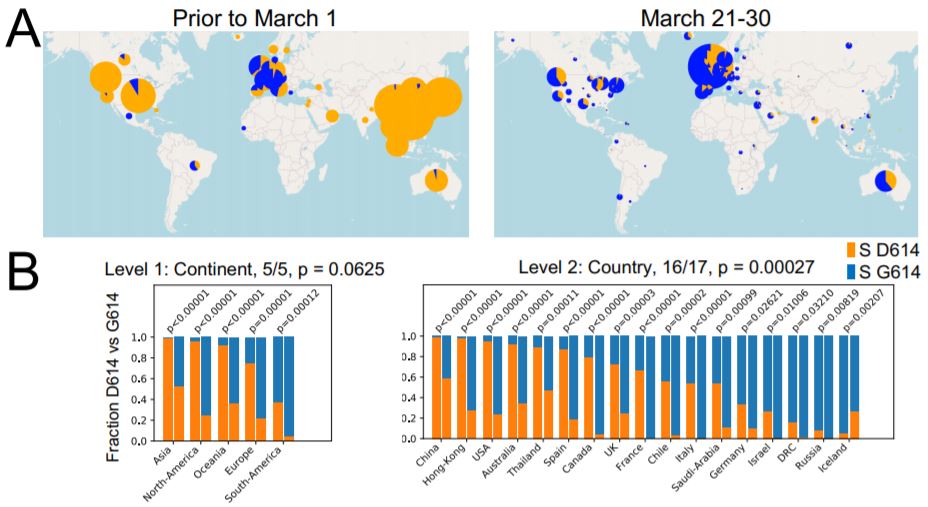
Distribuzioni delle varianti D614 (arancione) e G614 (blu) in Europa e nel mondo, con dettaglio per continente e per singolo Paese prima e dopo l'1 marzo. Korber et al 2020 Cell
La variante G614 fornirebbe un vantaggio evolutivo al virus poiché faciliterebbe il suo ingresso nelle cellule e dunque aumenterebbe la sua trasmissibilità, ma non solo: ad essa è associata una maggiore carica virale ma non necessariamente, scrivono gli autori, una maggiore severità della malattia. La virologa del San Raffaele Elisa Vicenzi, intervistata da Barbara Paknazar, ha sottolineato però che la maggior aggressività della variante ora più diffusa del virus è stata dimostrata in vitro ma non in vivo.
Ad oggi la pandemia da coronavirus è al massimo della sua espansione nel mondo. Il 10 luglio sono stati superati i 230.000 nuovi casi in un giorno, più di 71.000 di questi solo negli Stati Uniti, dove la California è tornata a sospendere molte attività avviandosi a un nuovo lockdown. Indizi che il virus abbia ridotto la sua aggressività ad oggi non ce ne sono, anzi, la mutazione della proteina Spike individuata dallo studio su Cell renderebbe il virus più aggressivo nel senso di più facilmente trasmissibile, senza però necessariamente provocare forme più gravi della malattia. Se l’Italia è riuscita ad abbassare fino a poche centinaia di nuovi casi giornalieri la curva dei contagi è merito delle misure restrittive messe in atto nei mesi di marzo, aprile e maggio. Che la genetica nostra o del virus ci abbia dato una mano in questa impresa ad oggi non risulta.


